
Defense mechanisms designed for the body’s immediate response to danger can, in critical cases, transform into destructive factors. Recent research reveals that during a myocardial infarction (heart attack), an overactive response from the nervous system—rather than isolating the damaged focus—becomes the primary driver for exacerbating the pathological state.
The process begins with the excitation of specialized sensory neurons (TRPV1) located within the heart. These nerve endings instantaneously perceive tissue damage and oxygen deprivation (hypoxia). They immediately transmit an alarm signal to the paraventricular nucleus (PVN) of the hypothalamus in the brain. The brain interprets this information as a critical threat and, in response, activates the superior cervical ganglia (SCG) located in the neck area.
Inflammatory cytokines (specifically IL-1β) released from these stimulated nerve nodes travel through nerve fibers back toward the heart. This process triggers a powerful inflammatory reaction within the damaged myocardial focus. Interestingly, when IL-1β was injected into the cervical nodes of healthy mice, they developed infarction-like complications and deterioration of cardiac function, while the brain’s PVN remained active. This confirmed the direction of information flow: the signal travels from the heart to the brain first, and only then reaches the cervical nerve nodes.
This so-called “toxic cycle” significantly accelerates the scarring of heart tissue (fibrosis). Consequently, the infarct zone expands, causing the heart to gradually lose its pumping function and leading to a state of decompensated heart failure.
Dysfunction of the Heart-Brain Axis in the Post-Infarct Period
The heart and brain maintain continuous communication through vagal sensory pathways, autonomic nerves, and the exchange of immune cells. During a myocardial infarction, the activity of these communication channels increases pathologically.
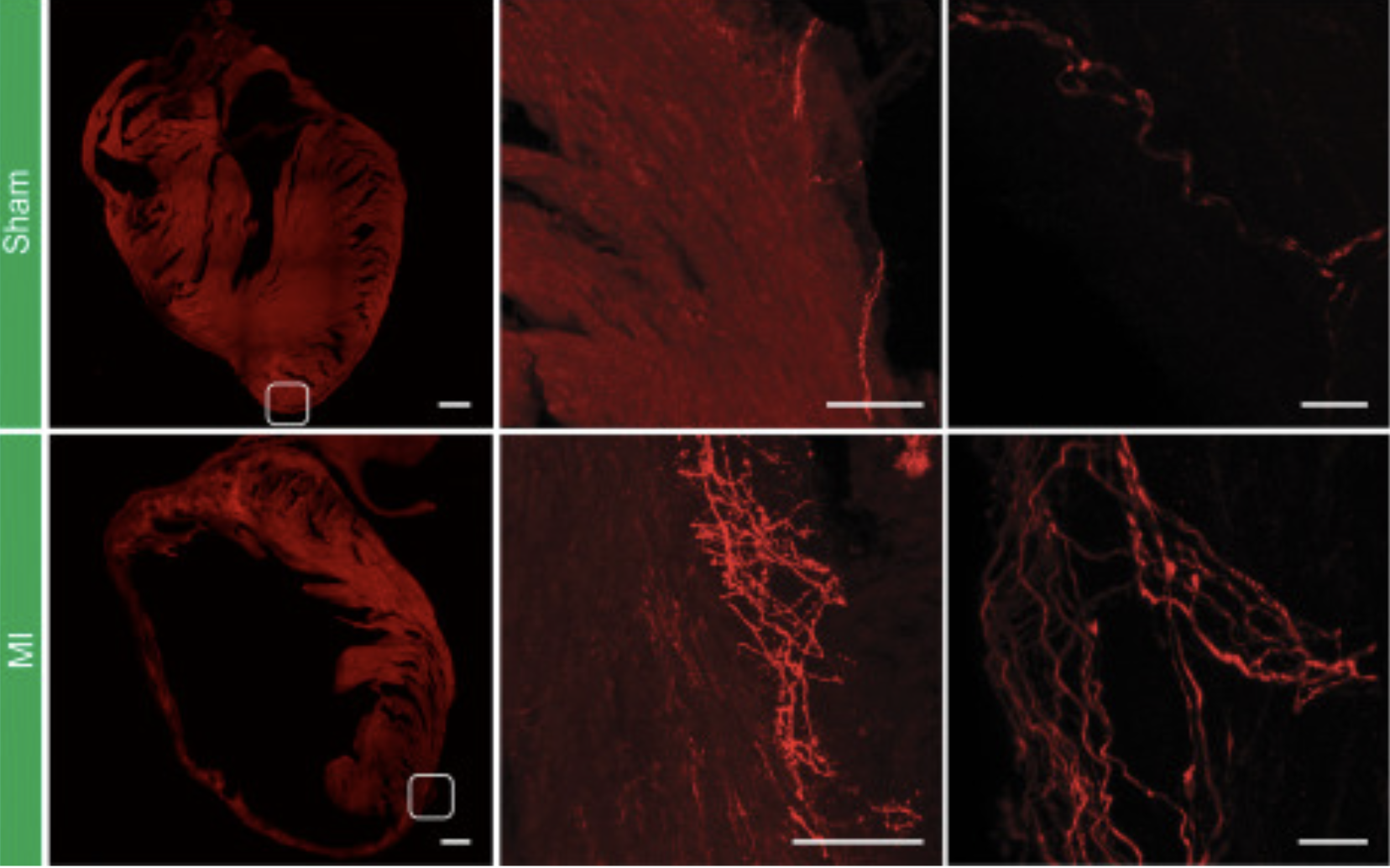
Using advanced genetic methods such as single-nucleus RNA sequencing (snRNA-seq), researchers explored these nerve nodes at a cellular level. They discovered that TRPV1 neurons represent a distinct group, qualitatively different from neurons that perceive only mechanical pressure. After a heart attack, the number of these labeled neurons increases significantly. Simultaneously, the network of nerve fibers around the damaged zone expands substantially.
Immunohistochemical analysis confirmed a sharp increase in nerve fibers in hearts that had undergone infarction. Using innovative tissue visualization methods that render biological material transparent, scientists were able to see the arrangement of these new fibers. They discovered that the nerves tightly encase the damaged area, almost like a protective layer. Thus, a heart attack forces the body to “re-wire” an additional sensory network within the ventricles.
Blocking Sensory Neurons and Restoring Cardiac Function
Scientists utilized a specific substance (resiniferatoxin) to deactivate (“ablate”) the TRPV1 nerve fibers. Two weeks after the infarction, this procedure partially restored the heart’s blood-pumping ability (ejection fraction). Electrocardiograms (EKG) showed that cardiac electrical impulses remained within the normal range in the nerve-ablated mice.
Echocardiographic examination confirmed that the size and volume of the heart’s ventricles approached the values of a healthy organ. As a result of this protective procedure, markers for blood vessel growth and cell restoration (CD31, VEGF) increased significantly. A drop in blood troponin levels indicated less damage to the heart muscle.
Laboratory testing of the tissues revealed that the infarct zone and the size of the scar were sharply reduced. Furthermore, the weight, temperature, and blood pressure of the mice remained unchanged, confirming the high precision of the intervention.
Cellular “Reprogramming”
On the third day post-infarction, researchers examined the nuclei of 126,734 cells in detail. The study showed that after the deactivation of the nerves, the proportion of major cell types remained unchanged, but their internal states were qualitatively transformed:
Endothelial Cells: Divided into eight subgroups, with a significant increase in cells capable of proliferation (reproduction).
Fibroblasts: Inflammatory processes within connective tissue cells decreased.
Macrophages: Switched to a “repair mode.”
Cardiomyocytes: Blocking the nerves restored the balance between damaged and healthy zones through specific genes, normalizing contractile ability, cellular connections, and electrical conductivity.
Research Limitations and Future Directions
Despite the achieved results, several questions remain unanswered. While IL-1β and TNF-α play major roles in inflammatory processes, other immune mediators also influence the heart’s response to infarction. Blocking nerve signals also alters the body’s general immune state, which requires further in-depth study.
Future attention will be devoted to the roles of T-cells and other cytokines. The exact mechanisms by which nerve impulses transition into sympathetic activation, including neurotransmitter release pathways, are not yet fully clear. Furthermore, TRPV1 neurons are also found in the dorsal root ganglia (DRG) of the spinal cord, whose role was not directly tested in this study.
Additionally, the heart is primarily supplied with sympathetic impulses by the stellate ganglia. Refining genetic technologies will allow for a more detailed study of the functional connections of these neural centers. Selective blocking of target nerve structures offers the possibility of developing individual therapeutic strategies, though clinical trials are necessary before these neuroimmune findings can be implemented in practice.
Source: Cell
